 Français
Français
 English
English
Les maladies
Les immunodéficiences primaires (DIP) regroupent plus de 400 maladies monogéniques rares qui affectent le développement, la fonction ou la régulation de la réponse immunitaire. Dans le monde, les DIP touchent 500 000 patients et, parmi ceux-ci, 62 % ont moins de 19 ans, tandis que plus de 25 % présentent une auto-immunité et une inflammation. Les DIPs associées à l'auto-immunité/inflammation présentent une expression variable, ce qui signifie qu'une mutation dans un gène peut entraîner différents phénotypes et qu'un phénotype peut être la conséquence de différents gènes affectés (figure 1).
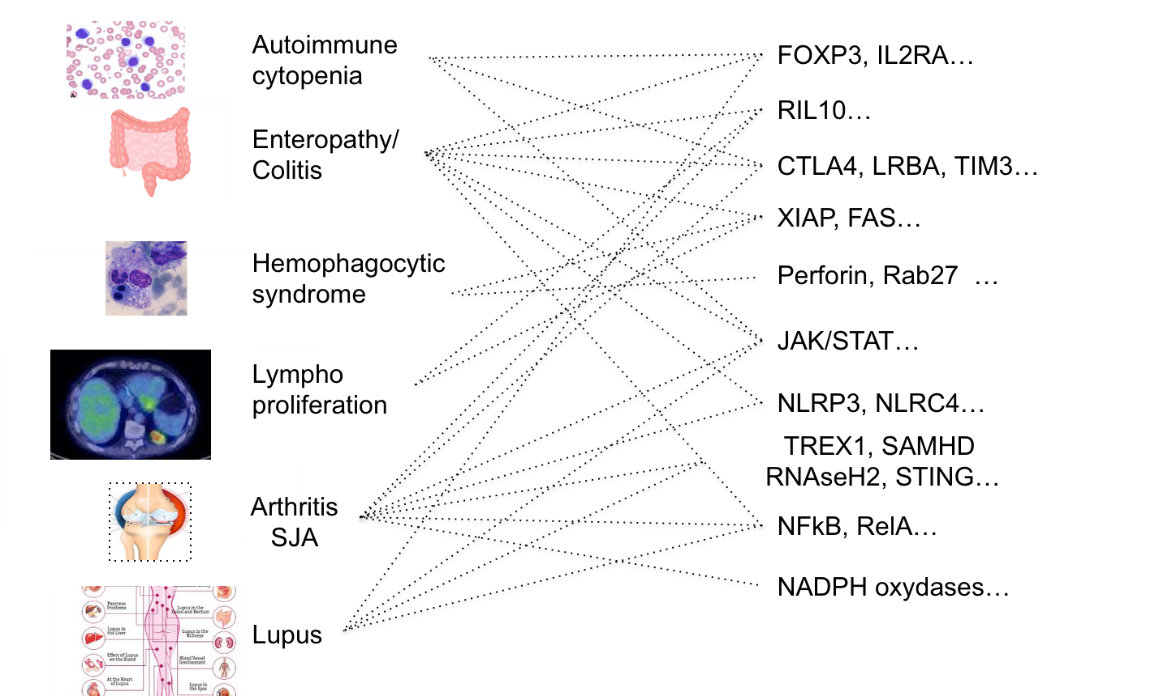
Figure 1. Types d'immunodéficiences primaires et les gènes identifiés qui sont affectés (de gauche à droite). Gènes affectés et types d'immunodéficiences primaires qui peuvent survenir (de droite à gauche).
Quelles maladies seront étudiées par le programme ?
-
Syndrome lymphoprolifératif avec autoimmunité (ALPS): Trouble héréditaire, d'apparition précoce (en moyenne <5 ans), dans lequel l'organisme ne peut pas réguler correctement le nombre de lymphocytes (cellules immunitaires), ce qui entraîne un nombre anormalement élevé de ces cellules dans le sang et dans les organes lymphoïdes secondaires (lymphoprolifération). Cette accumulation provoque l'hypertrophie des ganglions lymphatiques (lymphadénopathie), du foie (hépatomégalie) et de la rate (splénomégalie). Des manifestations auto-immunes sont observées chez plus des deux tiers des patients atteints d'ALPS, le plus souvent sous forme de cytopénies auto-immunes (anémie hémolytique, thrombocytopénie et neutropénie).
-
Cytopénie auto-immune: Groupe connexe de troubles dans lesquels les cellules hématopoïétiques différenciées sont détruites par le système immunitaire. La maladie monolignée est caractérisée par la production d'auto-anticorps contre les globules rouges (anémie hémolytique auto-immune [AIHA]), les plaquettes (thrombocytopénie auto-immune [ITP]) et les neutrophiles (neutropénie auto-immune [AIN]) alors que la maladie multilignée peut inclure diverses combinaisons de ces conditions.
-
Entéropathie - Maladies inflammatoires de l'intestin: Troubles impliquant une inflammation chronique du tube digestif. Ils comprennent la colite ulcéreuse, qui provoque une inflammation et des ulcères le long de la paroi du côlon et du rectum, et la maladie de Crohn, qui se caractérise par une inflammation à la fois de la paroi et des couches plus profondes du tube digestif. Elles partagent des symptômes tels que la diarrhée, les saignements rectaux, les douleurs abdominales, la fatigue et la perte de poids. Les causes restent inconnues.
-
Lupus érythémateux systémique (SLE): Maladie auto-immune dans laquelle le système immunitaire de l'organisme s'attaque par erreur à des tissus sains, provoquant une inflammation généralisée et des lésions tissulaires. C'est le type de lupus le plus courant. Il affecte la peau, les articulations, les reins, le cerveau et d'autres organes. Il n'y a pas de causes connues pour le SLE, mais des facteurs environnementaux, génétiques et hormonaux semblent être liés.
-
Arthrite juvénile idiopathique (AJI): Regroupe différents types d'AJI qui sont des maladies auto-immunes et inflammatoires est le type d'arthrite le plus fréquent chez les enfants et les adolescents. Elle provoque généralement des douleurs articulaires et des inflammations au niveau des mains, des genoux, des chevilles, des coudes et/ou des poignets. Ses causes sont inconnues.
-
Lymphistiocytose hémophagocytaire familiale (LHF): C'est un état dans lequel l'organisme fabrique trop de cellules immunitaires activées (macrophages et lymphocytes) qui provoquent de la fièvre, une hypertrophie du foie ou de la rate, une cytopénie (diminution du nombre de cellules sanguines) et des anomalies neurologiques. Elle peut avoir des causes génétiques ou non génétiques, qui distinguent l'HLH héritée de l'HLH acquise.